Publications
Selected Work by Topic
Computational Design and Engineering of Photoenzymes
Certain natural enzymes can be repurposed by light and carry out non-natural reactions. These enzymes can be tuned through protein engineering to carry out reactions that are challenging for other types of catalysts. We develop design strategies and predictive tools based on first-principles simulations to advance the emerging field of photoenzymatic catalysis.
Jose M. Carceller^, Bhumika Jayee^, Claire G. Page, Daniel G. Oblinsky, Nithin Chintala, Gustavo Mondragón-Solórzano, Jingzhe Cao, Zayed Alassad, Zheyu Zhang, Nathaniel White, Gregory D. Scholes*, Sijia S. Dong*, Todd K. Hyster*. Engineering a Photoenzyme to Use Red Light. DOI:10.26434/chemrxiv-2024-cjs5j
Accelerating Quantum Chemistry Simulations of Molecules and Condensed Systems Using Machine Learning and Automation
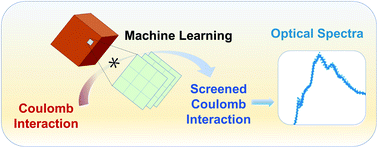
Accurate and efficient predictions of absorption spectra of molecules and solids are essential for the understanding and rational design of broad classes of materials, including photo-absorbers in solar and photo-electrochemical cells and defective insulators and semiconductors hosting optically addressable spin-defects. The prediction of absorption spectra at finite temperature is needed to directly compare with experimental observables. We have developed an approach to improve the efficiency of first principles calculations of absorption spectra of complex materials at finite temperature, based on the solution of the Bethe-Salpeter equation (BSE) and machine-learning techniques.
We demonstrate that methods based on convolution can be efficiently used to compute the screened Coulomb interaction, a key component of BSE calculations, with computational gains of one to two orders of magnitude for systems with 50 to 500 atoms, including liquids, solids, nanostructures, and solid/liquid interfaces. Importantly, our approach yields interpretable machine learning results, from which model dielectric functions may be derived. These model functions may be used not only in the BSE but also in developing functionals for time-dependent density functional theory (TDDFT) calculations, for both homogeneous and heterogeneous systems. Our work provides a strategy to combine machine learning with electronic structure calculations to accelerate the first principles simulations of excited-state properties.
Sijia S. Dong, Marco Govoni, Giulia Galli. Machine Learning Dielectric Screening for the Simulation of Excited State Properties of Molecules and Materials. Chem. Sci., 2021, 12, 4970-4980. (Link)
Multireference (strongly correlated) systems are ubiquitous in nature and in new generations of materials. For example, accurate description of photochemistry and transition metal spin states often requires the proper treatment of strong correlation. To study such systems, it is common to use multireference methods. However, the procedure for selecting an active space to obtain the multiconfigurational wave functions is often nonsystematic. We have developed several approaches to systematically select the active space to generate reasonable reference wave functions for complete active space 2nd-order perturbation theory and multiconfiguration pair-density functional theory to accurately predict the excited states of various molecules.
Benjamin W. Kaufold, Nithin Chintala, Pratima Pandeya, Sijia S. Dong*. Automated Active Space Selection with Dipole Moments. J. Chem. Theory Comput., 2023, 19, 9, 2469–2483. (Link)
Excited States of Biomolecules and Materials
The correct description of the excited states of chemical systems is critical for understanding and designing light-harvesting molecules and materials. However, many such systems are multireference (strongly-correlated) in nature, and the widely used Kohn-Sham density functional theory (KS-DFT) cannot describe such systems well. Here, we use multiconfiguration pair-density functional theory (MC-PDFT) and other electronic structure methods to study the photochemistry of biomolecules and building blocks of functional materials.

Conjugated organic molecules comprise a large class of molecules that play important roles in nature and in chemical applications such as photosynthesis, vision, light-driven molecular motors, and organic electronics. Butadiene is the simplest case of a conjugated organic molecule, and has been studied extensively. However, the inferences about the nature of its lowest excited states were still inconclusive. In this work, we resolve this issue. We demonstrate that a combined analysis of the orbital second moments, state second moments, and MC-PDFT energy components is a very useful approach in determining excited-state characteristics and assigning states, and we show that the level of diffuseness of basis sets that should be used follows a Goldilocks principle.
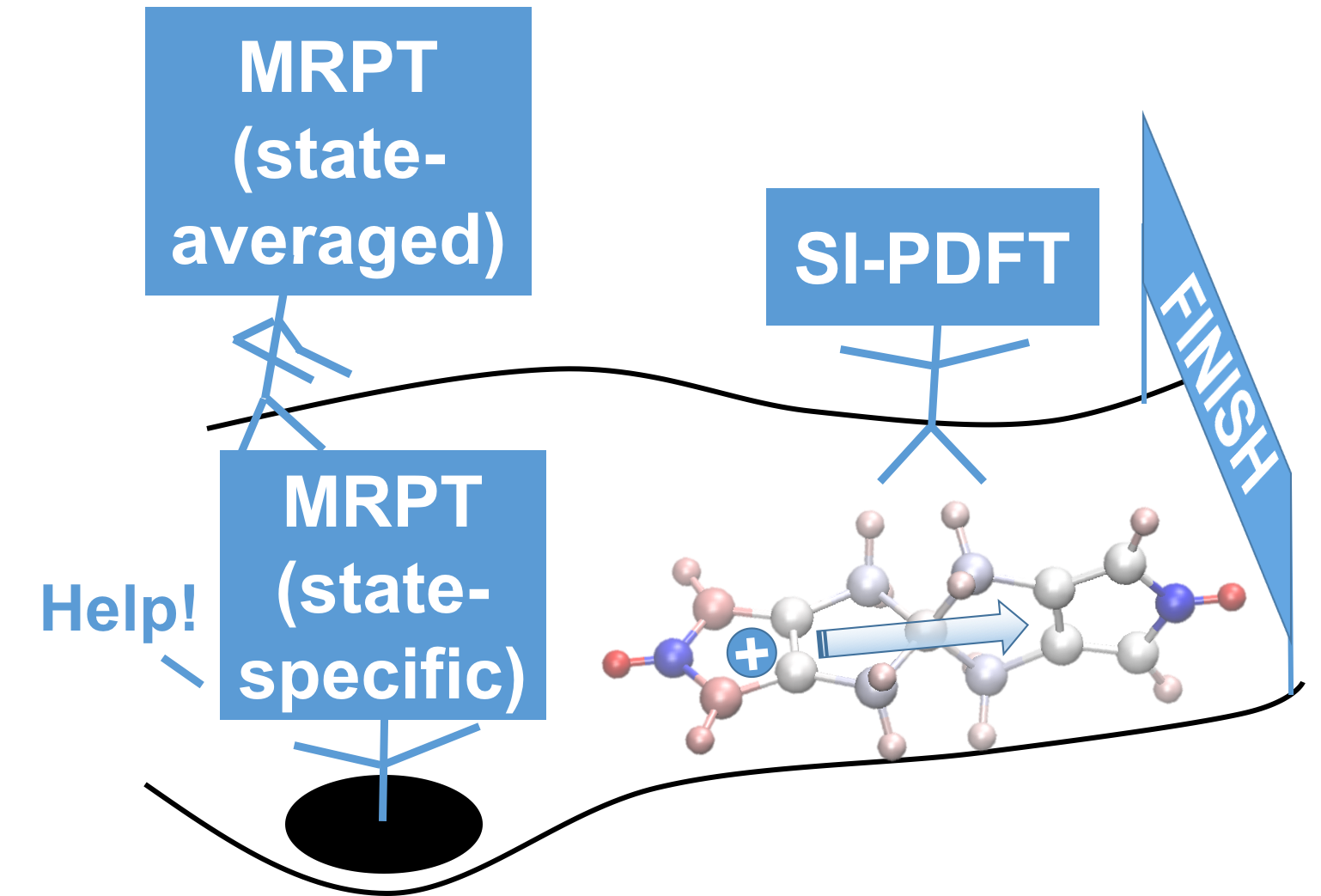
Charge transfer (CT) is a common chemical phenomenon and is critical in modern technology. Among CT systems, mixed-valence compounds with strong couplings between electronic states are one of the most challenging types for electronic structure theory. We show that the state-interaction (SI) flavor of MC-PDFT is free from the unphysical behavior of previously tested second-order multireference perturbation theory methods (MRPT2, a class of commonly-used multireference methods that are much more computationally expensive) for a prototype mixed-valence compound near the avoided crossing. This is very encouraging because of the much lower cost in applying SI-PDFT to large or complex systems.
Sijia S. Dong^, Benchen Huang^, Laura Gagliardi, Donald G. Truhlar. State-Interaction Pair-Density Functional Theory Can Accurately Describe a Spiro Mixed-Valence Compound. J. Phys. Chem. A, 2019, 123, 10, 2100-2106. (Published as part of The Journal of Physical Chemistry virtual special issue “Leo Radom Festschrift”.) (Link)
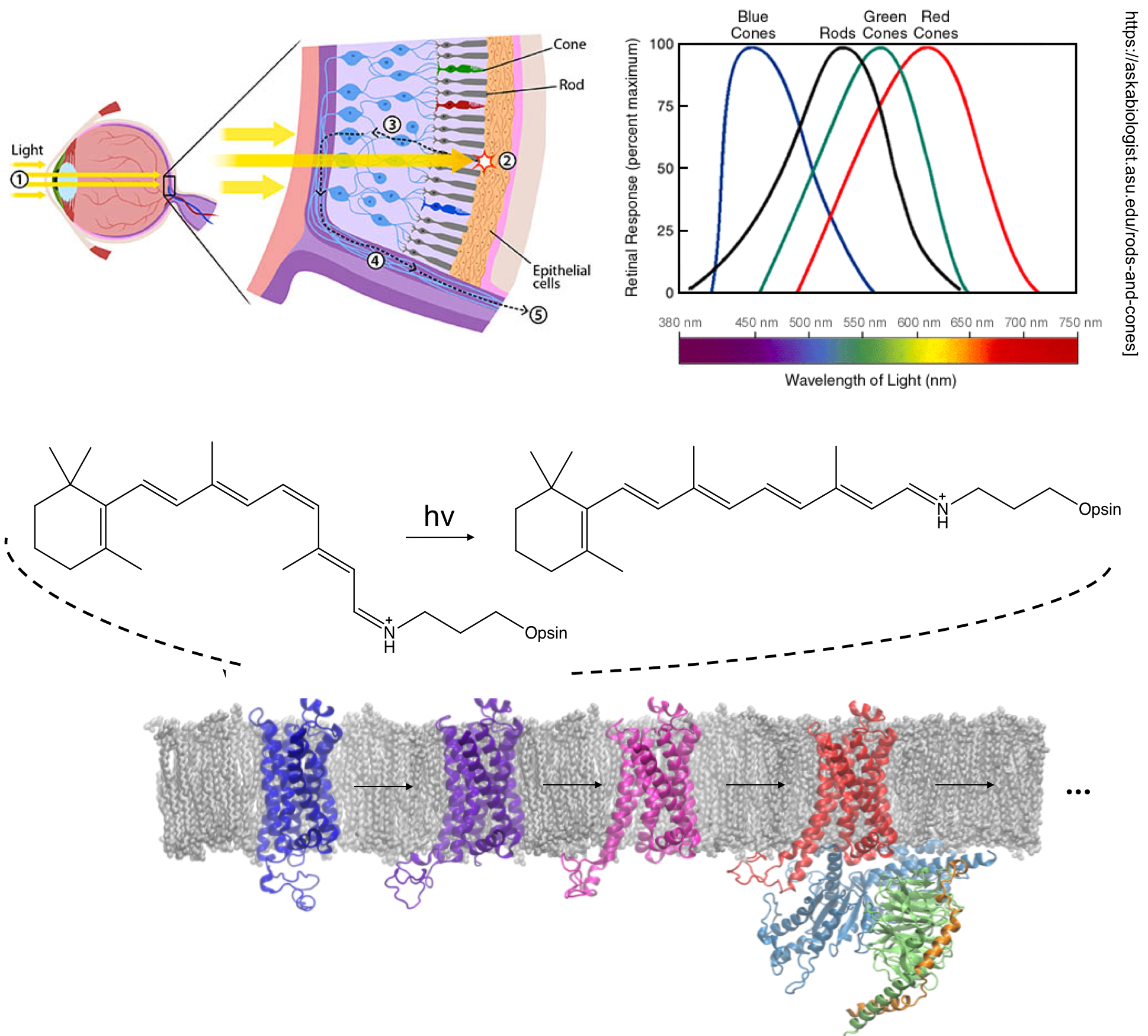
The retinal molecule has been an important prototype target of photochemistry research for decades. It is covalently bound to a type of G protein-coupled receptors (GPCRs), opsins, which are responsible for vision. The retinal ligand undergoes cis-trans isomerization upon light, which triggers conformational changes in the opsin protein from the “inactive state” to the “active state”. The activated opsin is involved in signal transduction in cells, which is eventually transformed into visual signals in brains.
The absorption maximum of retinal is sensitive to the mutations in the protein. Therefore, it is essential to predict the absorption spectrum of retinal accurately. However, the presence of charge transfer excitations in this molecule and its analogues poses a challenge for KS-DFT, and the large size of the molecule makes multiconfigurational perturbation theory methods expensive. We have demonstrated that MC-PDFT provides an efficient way in predicting the vertical excitation energies of retinals. With this tool, we will be able to understand and design other chromophores important for biological and energy research.
Sijia S. Dong, Laura Gagliardi, Donald G. Truhlar. Excitation Spectra of Retinal by Multiconfiguration Pair-Density Functional Theory. Phys. Chem. Chem. Phys., 2018, 20, 7265-7276. (Link)
María del Carmen Marín, Luca De Vico, Sijia S. Dong, Laura Gagliardi, Donald G. Truhlar, Massimo Olivucci. Assessment of MC-PDFT Excitation Energies for a Set of QM/MM Models of Rhodopsins. J. Chem. Theory Comput., 2019, 15, 3, 1915-1923. (Link)
Conformational Landscape of Biological and Synthetic Macromolecules
Macromolecules have complex potential energy surfaces, and the sampling of their conformational landscape is necessary for predicting their properties. We combine first-principles simulations and data-driven methods to explore the conformational landscape of proteins, synthetic polymers, and other macromolecules.
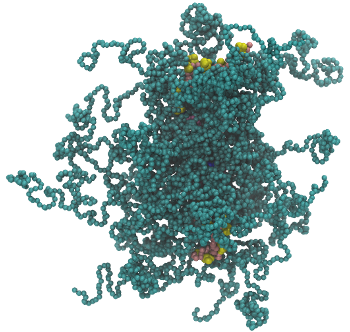
All-Atom and Coarse-Grained Modeling of Macromolecules to Inform Experimental Design Certain bottle-brush polymers are capable of delivering oligonucleotide therapeutics into cells. To rationally design these polymers for desired cellular uptake, a mechanistic understanding is necessary. My research group developed all-atom and coarse-grained force fields for these polymers to understand their morphology and their interactions with the cell membrane. We use simulations to inform the experimental design of these polymers.
Yuyan Wang, Dali Wang, Jiachen Lin, Zidi Lyu, Peiru Chen, Tingyu Sun, Mehrnaz Mojtabavi, Armin Vedadghavami, Zheyu Zhang, Ruimeng Wang, Lei Zhang, Christopher Park, Sijia S. Dong*, and Ke Zhang*. A Long-Circulating Vector for Aptamers Based upon Polyphosphodiester-Backboned Molecular Brushes. Angew. Chem. Int. Ed., 2022, 61, e202204576. (Link)
Dali Wang, Qiwei Wang, Yuyan Wang, Peiru Chen, Xueguang Lu, Fei Jia, Yehui Sun, Tingyu Sun, Lei Zhang, Fangyuan Che, Jialu He, Liming Lian, Gemma Morano, Michael Shen, Mengqi Ren, Sijia S. Dong, Jean Zhao, and Ke Zhang. Targeting oncogenic KRAS with molecular brush-conjugated antisense oligonucleotides. PNAS, 2022, 119 (29), e2113180119. (Link)
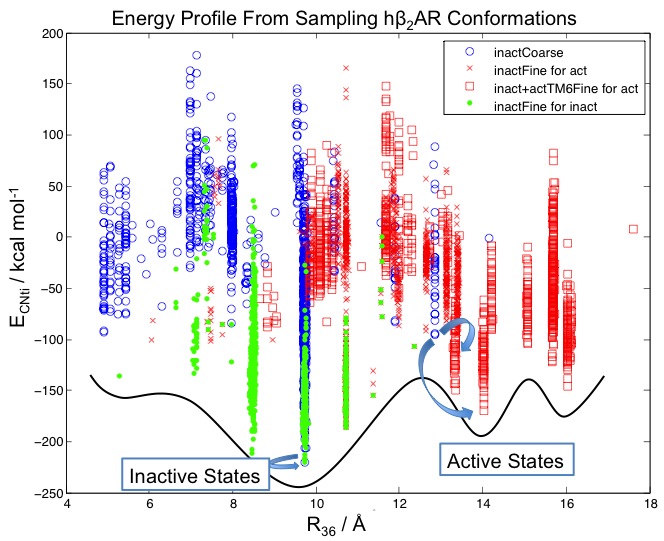
Protein Structure Prediction: One Sequence, Multiple Conformations G protein-coupled receptors play critical signaling functions for numerous cellular processes, making important targets for therapeutics. About 40% of drugs on the market target on these membrane proteins. However, developing such therapeutics is complicated because the activation of GPCRs that is integral to their function involves multiple distinct conformations along the pathway for activation, and obtaining these conformations experimentally is challenging.
To address this challenge, we developed a computational methodology, ActiveGEnSeMBLE, to sample and predict from first principles the multiple conformations along the activation coordinates of GPCRs. We select a handful of candidate conformations from trillions of possible conformations. We then subjected the predicted structures to molecular dynamics simulations and obtained a quantitative energy landscape that provides insights to GPCRs’ activation mechanisms not easily accessible by experiments. It was, we believe, the first quantitative energy profile for GPCR activation consistent with the qualitative profile deduced from experiments. We have applied the method to a GPCR without an experimental structure and have made experimentally testable hypotheses for drug discovery.
Sijia S. Dong, William A. Goddard III, Ravinder Abrol. Identifying Multiple Active Conformations in the G Protein-Coupled Receptor Activation Landscape Using Computational Methods. In Methods in Cell Biology. 2017; vol. 142, pp. 173-186.(Link)
Sijia S. Dong, William A. Goddard III, Ravinder Abrol. Conformational and Thermodynamic Landscape of GPCR Activation From Theory and Computation. Biophys. J., 2016, 110 (12), 2618-2629. (Link)
Sijia S. Dong, Ravinder Abrol, William A. Goddard III. The Predicted Ensemble of Low Energy Conformations of Human Somatostatin Receptor Subtype 5 and the Binding of Antagonists. ChemMedChem, 2015, 10 (4), 650–661. (Link)
Inorganic Chemistry and Catalysis
In close collaboration with experimental chemists, we leveraged the power of computational chemistry to provide insights into the electronic structures of transition metal complexes and the reaction mechanisms of transition metal catalysts.
Benjamin S. Rich, Noah B. Bissonnette, Alejandra Duran Balsa, Mulan Yang, Hope Meikle, Nithin Chintala, Sijia S. Dong* and Rein U. Kirss*. Mechanism of Halide Exchange in Reactions of CpRu(PPh3)2Cl with Haloalkanes. New J. Chem., 2022, 46, 6603-6608. (Link)
Xin-Yuan Liu^, Zhen Guo^, Sijia S. Dong^, Xiao-Hua Li and Chi-Ming Che. Highly Efficient and Diastereoselective Gold (I)-Catalyzed Synthesis of Tertiary Amines from Secondary Amines and Alkynes: Substrate Scope and Mechanistic Insights. Chemistry-A European Journal, 2011, 17 (46), 12932–12945. (Link)
Sijia S. Dong, Robert J. Nielsen, Joshua H. Palmer, Harry B. Gray, Zeev Gross, Siddharth Dasgupta, William A. Goddard III. Electronic Structures of Group 9 Metallocorroles with Axial Ammines. Inorganic Chemistry, 2011, 50 (3), 764–770. (Link)
Large-Scale Non-Adiabatic Electron Dynamics
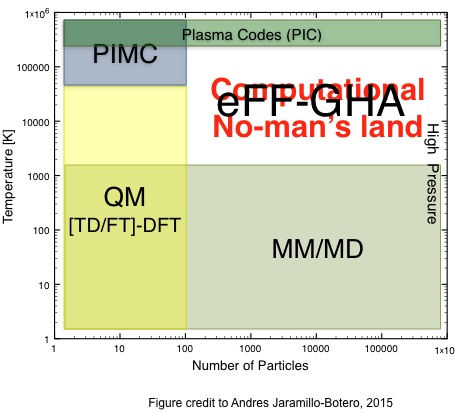
We develop a model, Gaussian Hartree Approximated Quantum Mechanics (GHA-QM) With Angular Momentum Projected Effective coRe potEntial (AMPERE), for doing non-adiabatic electron dynamics. GHA-QM is based on the electron force field (eFF) concept, which solves the time-dependent Schrödinger equation with the electrons approximated as Gaussian wavepackets and nuclei point charges. An introduction to eFF is here. The eFF method has been applied to modeling systems and processes involving excited electron dynamics such as warm dense hydrogen, Auger process in hydrocarbon nanoparticles, and electronic effects in brittle fracture of silicon.
Hai Xiao, Sijia S. Dong, Andres Jaramillo-Botero, William A. Goddard III. Large-Scale Quantum Non-Adiabatic Electron Dynamics Using Gaussian Hartree Approximated Quantum Mechanics With Angular Momentum Projected Effective Core Potential.
Preprints
Jose M. Carceller^, Bhumika Jayee^, Claire G. Page, Daniel G. Oblinsky, Nithin Chintala, Gustavo Mondragón-Solórzano, Jingzhe Cao, Zayed Alassad, Zheyu Zhang, Nathaniel White, Gregory D. Scholes*, Sijia S. Dong*, Todd K. Hyster*. Engineering a Photoenzyme to Use Red Light. DOI:10.26434/chemrxiv-2024-cjs5j
Journal Articles
Katherine Carney, Lauren Polito, Kamilya Reid, Surbhi Srinivas, Gabrielle Blake, Nithin Chintala, Sijia S. Dong* and Rein Kirss*. Kinetics and Mechanism of Halide Exchange in Reactions of CpRu(PPh3)2Cl with Alkyl Halides: Evidence for Radical Pairs. New J. Chem., 2023,47, 21404-21410. (Link)
Benjamin W. Kaufold, Nithin Chintala, Pratima Pandeya, Sijia S. Dong*. Automated Active Space Selection with Dipole Moments. J. Chem. Theory Comput., 2023, 19, 9, 2469–2483. (Link)
Giovanni Li Manni, Ignacio Fernández Galván, …, Sijia S. Dong, et al. The OpenMolcas Web: A Community-Driven Approach to Advancing Computational Chemistry. J. Chem. Theory Comput., 2023, 19, 20, 6933–6991. (Link)
Lei Zhang^, Yuyan Wang^, Peiru Chen, Dali Wang, Tingyu Sun, Zheyu Zhang, Ruimeng Wang, Xi Kang, Yang Fang, Hao Lu, Jiansong Cai, Mengqi Ren, Sijia S. Dong*, and Ke Zhang*. A mechanistic study on the cellular uptake, intracellular trafficking, and antisense gene regulation of bottlebrush polymer-conjugated oligonucleotides. RSC Chem. Biol., 2023, 4, 138-145. (Link)
Yuyan Wang, Dali Wang, Jiachen Lin, Zidi Lyu, Peiru Chen, Tingyu Sun, Mehrnaz Mojtabavi, Armin Vedadghavami, Zheyu Zhang, Ruimeng Wang, Lei Zhang, Christopher Park, Sijia S. Dong*, and Ke Zhang*. A Long-Circulating Vector for Aptamers Based upon Polyphosphodiester-Backboned Molecular Brushes. Angew. Chem. Int. Ed., 2022, 61, e202204576. (Link)
Dali Wang, Qiwei Wang, Yuyan Wang, Peiru Chen, Xueguang Lu, Fei Jia, Yehui Sun, Tingyu Sun, Lei Zhang, Fangyuan Che, Jialu He, Liming Lian, Gemma Morano, Michael Shen, Mengqi Ren, Sijia S. Dong, Jean Zhao, and Ke Zhang. Targeting oncogenic KRAS with molecular brush-conjugated antisense oligonucleotides. PNAS, 2022, 119 (29), e2113180119. (Link)
Benjamin S. Rich, Noah B. Bissonnette, Alejandra Duran Balsa, Mulan Yang, Hope Meikle, Nithin Chintala, Sijia S. Dong* and Rein U. Kirss*. Mechanism of Halide Exchange in Reactions of CpRu(PPh3)2Cl with Haloalkanes. New J. Chem., 2022, 46, 6603-6608. (Link)
Sijia S. Dong, Marco Govoni, Giulia Galli. Machine Learning Dielectric Screening for the Simulation of Excited State Properties of Molecules and Materials. Chem. Sci., 2021, 12, 4970-4980. (Editor’s Choice – Graeme Day, Materials & Energy in Chemical Science - most popular articles 2021) (Link)
Before 2021
Ignacio Fdez. Galván, Morgane Vacher, …, Sijia S. Dong, et al. OpenMolcas: From Source Code to Insight. J. Chem. Theory Comput., 2019, 15, 11, 5925-5964. (Link)
Sijia S. Dong, Laura Gagliardi, Donald G. Truhlar. Nature of the \(1^1B_{u}\) and \(2^1A_{g}\) Excited States of Butadiene and the Goldilocks Principle of Basis Set Diffuseness. J. Chem. Theory Comput., 2019, 15, 8, 4591-4601. (Link)
Sijia S. Dong^, Benchen Huang^, Laura Gagliardi, Donald G. Truhlar. State-Interaction Pair-Density Functional Theory Can Accurately Describe a Spiro Mixed-Valence Compound. J. Phys. Chem. A, 2019, 123, 10, 2100-2106. (Published as part of The Journal of Physical Chemistry virtual special issue “Leo Radom Festschrift”.) (Link)
María del Carmen Marín, Luca De Vico, Sijia S. Dong, Laura Gagliardi, Donald G. Truhlar, Massimo Olivucci. Assessment of MC-PDFT Excitation Energies for a Set of QM/MM Models of Rhodopsins. J. Chem. Theory Comput., 2019, 15, 3, 1915-1923. (Link)
Yinan Shu, Sijia S. Dong, Kelsey A. Parker, Junwei L. Bao, Linyao Zhang, Donald G. Truhlar. Extended Hamiltonian Molecular Dynamics: Semiclassical Trajectories with Improved Maintenance of Zero Point Energy. Phys. Chem. Chem. Phys., 2018, 20, 30209-30218. (Highlighted as a 2018 PCCP HOT Article.) (Link)
Jie J. Bao, Sijia S. Dong, Laura Gagliardi, and Donald G. Truhlar. Automatic Selection of an Active Space for Calculating Electronic Excitation Spectra by MS-CASPT2 or MC-PDFT. J. Chem. Theory Comput., 2018, 14 (4), 2017–2025. (Link)
Sijia S. Dong, Laura Gagliardi, Donald G. Truhlar. Excitation Spectra of Retinal by Multiconfiguration Pair-Density Functional Theory. Phys. Chem. Chem. Phys., 2018, 20, 7265-7276. (Link)
Sijia S. Dong, William A. Goddard III, Ravinder Abrol. Conformational and Thermodynamic Landscape of GPCR Activation from Theory and Computation. Biophys. J., 2016, 110 (12), 2618-2629. (Link)
Ruijie D. Teo^, Sijia S. Dong^, Zeev Gross, Harry B. Gray, William A. Goddard III. Computational Predictions of Corroles as a Class of Hsp90 Inhibitors. Molecular BioSystems, 2015, 11, 2907-2914. (Link)
Sijia S. Dong, Ravinder Abrol, William A. Goddard III. The Predicted Ensemble of Low Energy Conformations of Human Somatostatin Receptor Subtype 5 and the Binding of Antagonists. ChemMedChem, 2015, 10 (4), 650–661. (Link)
Xin-Yuan Liu^, Zhen Guo^, Sijia S. Dong^, Xiao-Hua Li and Chi-Ming Che. Highly Efficient and Diastereoselective Gold (I)-Catalyzed Synthesis of Tertiary Amines from Secondary Amines and Alkynes: Substrate Scope and Mechanistic Insights. Chemistry-A European Journal, 2011, 17 (46), 12932–12945. (Link)
Sijia S. Dong, Robert J. Nielsen, Joshua H. Palmer, Harry B. Gray, Zeev Gross, Siddharth Dasgupta, William A. Goddard III. Electronic Structures of Group 9 Metallocorroles with Axial Ammines. Inorganic Chemistry, 2011, 50 (3), 764–770. (Link)
Book Chapter
Sijia S. Dong, William A. Goddard III, Ravinder Abrol. Identifying Multiple Active Conformations in the G Protein-Coupled Receptor Activation Landscape Using Computational Methods. In Methods in Cell Biology. 2017; vol. 142, pp. 173-186.(Link)
^ denotes equal contribution